
Синдром драйвер что это
Синдром Драве: причины, клиника, диагностика, лечение
Впервые синдром Драве был описан Charlotte Dravet как тяжелая миоклоническая эпилепсия младенцев. Хотя синдром часто включает в себя миоклонические феномены, они редко бывают первым проявлением заболевания. Полученные в ходе развития современных методов молекулярной генетики данные подтвердили основанные на клинических наблюдениях предположения (Veggiotti et al., 2001) о том, что синдром Драве является частью спектра тяжелых эпилепсий, клиническая картина которых может дополняться генерализованными тонико-клоническими припадками, фокальными припадками, атипичными абсансами и миоклониями.
Синдром Драве считается наиболее тяжелым расстройством спектра эпилептических синдромов, описываемых как «GEFS+» (Generalized Epilepsy with Febrile Seizures plus — генерализованная эпилепсия с фебрильными припадками плюс).
Более 80% случаев синдрома Драве вызвано вновь возникшей мутацией гена натриевого канала SCN1A (Claes et al., 2001). Описано более 100 новых мутаций (Mulley et al., 2005), наиболее часто «усеченные» мутации, но также сплайс-сайт мутации, делеции и миссенс-мутации. В оставшихся 20% случаев мутации гена SCNA1 отсутствуют. Сообщалось о двух семейных случаях мутаций гена GABRG2. Недавно Depienne et al. (2006) сообщили о соматическом и половом клеточном мозаицизме у родителей — бессимптомных носителей генов в двух семьях с двумя и более случаями синдрома Драве.
Начало, обычно в возрасте 4-10 месяцев, характеризуется клоническими припадками, часто односторонними и длительными, в 75% случаев возникающими при высокой температуре. Такие припадки неоднократно рецидивируют через короткие интервалы (обычно менее двух месяцев). В начале заболевания ребенок развивается нормально. В течение второго или третьего года жизни возникают другие типы припадков, включая парциальные припадки, атипичные абсансы, миоклонические судороги и эпизоды неконвульсивного статуса. В этот же период становится заметной задержка когнитивного развития.
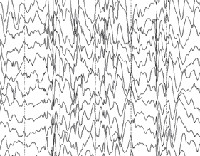
Часто выявляются симптомы поражения пирамидного тракта и атаксия. Несмотря на часто повторяющиеся припадки, в течение первых месяцев заболевания на интериктальной ЭЭГ обычно не обнаруживается патологических изменений. Начиная со второго года жизни, на ЭЭГ отсутствуют медленные комплексы спайк-волна, появляются быстрые комплексы спайк-волна, часто вместе с мультифокальными пиками. Фоточувствительность имеется у 25% пациентов, и нередко наблюдается самопроизвольное начало припадка. Долгосрочный прогноз заболевания неблагоприятен. Сохраняются припадки в основном в виде тонико-клонических атак, а миоклонические припадки постепенно исчезают.
Во всех случаях присутствует умственная отсталость различной степени (Oguni et al., 2001b; Dravet и Bureau, 2005).

У некоторых пациентов генерализованные или односторонние клонические припадки, вызываемые небольшим подъемом температуры тела или даже горячей ванной, являются преобладающим или единственным видом припадков, тогда как короткие припадки могут не быть ведущим симптомом и могут не включать в себя миоклонические атаки, которые в любом случае редко являются выраженными проявлениями эпилепсии. Некоторые исследователи описывали подобные случаи как вариант миоклонической эпилепсии (Ogino et al., 1989; Watanabe et al., 1989), эпилепсии c геми-grand mal и медленными волнами с большой амплитудой на ЭЭГ (Doose, 1992) или как идиопатическую эпилепсию младенцев (Ebach et al., 2005).
Такие формы сгруппированы (Oguni et al., 2001а; Fukuma et al., 2004) в «пограничные случаи» тяжелой миоклонической эпилепсии. Течение при этом не отличается от типичных форм, хотя может быть менее тяжелым. Было показано, что некоторые из этих атипичных форм вызваны мутациями гена SCNA1, но часто это другие мутации, нежели те, которые вызывают типичные формы, особенно редко встречаются «усеченные» мутации (Claes et al., 2001; Fujivara et al., 2003; Fukumura et al., 2004). Тот же ген может быть ответственным за развитие и других эпилептических синдромов, включая эпилепсию с миоклонически-ми астатическими припадками и некоторые семейные случаи GEFS+ (Ebah et al, 2005).
Harkin et al. (2007) проанализировали 90 случаев «эпилептической энцефалопатии» с мутациями SCN1A: у 52 пациентов был типичный синдром Драве, у 25 — атипичные формы, у 6 была криптогенная генерализованная эпилепсия, у 4 — криптогенная фокальная эпилепсия, у 2 — миоклоническая-астатическая эпилепсия и у 1 —синдром Леннокса-Гасто, что демонстрирует варианты экспрессии гена.
Berkovic et al. (2006) выявили мутации SCN1A у 11 из 14 пациентов с так называемой вакцинальной энцефалопатией. Клинико-молекулярный корреляционный анализ выявил мутации в 8 случаях с фенотипом синдрома Драве, в 3 из 4 случаев пограничного синдрома Драве, и ни в одном из двух случаев синдрома Леннокса-Гасто. Эти данные, хотя и нуждаются в проверке, обеспечивают солидное альтернативное объяснение причинам возникновения подобных детских расстройств; эти случаи могут быть вызваны совершенно другими причинами, вне связи с вакцинацией, вопреки распространенному мнению.
Необходимо тщательно взвешивать взаимодействие стирипенто-ла с большим количеством других препаратов. При том, что полный контроль достигается в редких случаях, все же представляется, что эти комбинации уменьшают как частоту, так и длительность конвульсивных эпизодов. В прошлом вигабатрин давал хорошие результаты у пациентов с ослабленным миоклоническим компонентом. Наконец, применение топирамата дало обнадеживающие результаты в открытых исследованиях, вероятно, препарат должен назначаться в раннем периоде заболевания (Dravet и Bureau, 2005). Комбинация топирамата и стирипентола представляется простой и безопасной (Kroll-Seger et al„ 2006). Сообщалось, что ламотриджин вызывает ухудшение у маленьких детей с синдромом Драве (Guerrini et al, 1998b).
Синдром длительного раздавливания – это шокоподобное состояние, наступающее после длительного сдавления туловища, конечностей или их сегментов тяжелыми предметами. Проявляется болью, ухудшением состояния, отеком пораженных отделов тела, острой почечной недостаточностью. Без медицинской помощи пациенты погибают от ОПН, нарастающей интоксикации, легочной или сердечно-сосудистой недостаточности. Лечение включает дезинтоксикационную и плазмозаменяющую инфузионную терапию, экстракорпоральную гемокоррекцию, антибиотикотерапию, иссечение участков некроза или ампутацию раздавленной конечности.
МКБ-10

Общие сведения
Синдром длительного раздавливания (СДР), другие названия – травматический токсикоз, краш-синдром, синдром Байуотерса, миоренальный синдром – патологическое шокоподобное состояние, наступающее после длительного сдавления туловища, конечностей или их сегментов тяжелыми предметами. Краш-синдром развивается сразу после освобождения больного и восстановления крово- и лимфотока в пораженных частях тела. Сопровождается ухудшением общего состояния, развитием токсемии и острой почечной недостаточности, при большой площади поражения нередко заканчивается смертью пациента. В травматологии и ортопедии выделяют бытовую разновидность краш-синдрома – так называемый синдром позиционного сдавления (СПС), который развивается в результате длительного (более 8 часов) сдавливания частей тела во время неподвижного положения человека на твердой поверхности.
Причины СДР
Обычно синдром длительного раздавливания возникает у пострадавших во время оползней, землетрясений, обвалов в шахтах, строительных работ, дорожных аварий, заготовки леса, взрывах и разрушениях зданий в результате бомбардировки.
Синдром позиционного сдавления обычно выявляется у пациентов, которые на момент травмы находились в состоянии отравления снотворными препаратами, наркотического или алкогольного опьянения. Чаще страдают подвернутые под туловище верхние конечности. По причинам развития, симптомам и методам лечения синдром позиционного сдавления практически не отличается от синдрома длительного раздавливания, однако, обычно протекает более благоприятно вследствие меньшей площади поражения.
Патогенез
Возникновение синдрома длительного раздавливания обусловлено сочетанием трех факторов:
- болевого синдрома;
- массивной потери плазмы, обусловленной выходом жидкой части крови через стенки сосудов в поврежденные ткани;
- травматической токсемии (интоксикации организма продуктами тканевого распада).
Продолжительное болевое раздражение при краш-синдроме приводит к развитию травматического шока. Потеря плазмы становится причиной сгущения крови и вызывает тромбоз мелких сосудов. Травматическая токсемия при краш-синдроме развивается вследствие всасывания в кровь продуктов тканевого распада травмированных мышц. Сразу после освобождения конечности из поврежденных тканей в сосудистое русло поступает значительное количество ионов калия, которые могут вызвать аритмию, а в тяжелых случаях – прекращение работы легких и сердца.
В дальнейшем раздавленные мышечные ткани пациента с краш-синдромом теряют до 66% калия, 75% миоглобина, 75% фосфора и 70% креатинина. Продукты распада поступают в кровь, вызывая ацидоз и нарушения гемодинамики (в том числе – резкое сужение сосудов почечных клубочков). Миоглобин повреждает и закупоривает почечные канальцы. Все это приводит к развитию острой почечной недостаточности, угрожающей жизни больного краш-синдромом.
Классификация
По степени тяжести:
- Легкая форма краш-синдрома. Возникает при раздавливании сегментов конечности в течение 4 и менее часов.
- Среднетяжелая форма краш-синдрома. Развивается в результате раздавливания одной конечности в течение 4-6 часов. При своевременном начале лечения прогноз благоприятный.
- Тяжелая форма краш-синдрома. Возникает при раздавливании одной конечности в течение 6-8 часов. Сопровождается расстройствами гемодинамики и острой почечной недостаточностью. При своевременном начале лечения прогноз относительно благоприятный.
- Крайне тяжелая форма краш-синдрома. Развивается в результате раздавливания двух и более конечностей в течение 6 и более часов. Сопровождается тяжелым шоком. Прогноз неблагоприятный.
По клинической симптоматике:
- ранний период (с момента освобождения до 3 суток);
- токсический период (начинается на 4-5 сутки);
- период поздних осложнений (развивается, спустя 20-30 суток с момента травмы).
Симптомы СДР
Сразу после устранения сдавления общее состояние пострадавшего улучшается. Пациента с синдромом длительного раздавливания беспокоит боль и ограничение движений в раздавленной конечности. В течение первых часов после освобождения постепенно нарастает отек пострадавшего участка, который становится плотным, деревянистым. На коже конечности образуются пузыри с серозно-геморрагическим содержимым. При осмотре поврежденной части тела выявляется ослабление пульсации артерий, снижение чувствительности и местной температуры.
Нарастает общая симптоматика. Состояние пострадавшего с краш-синдромом ухудшается. После короткого периода возбуждения пациент становится вялым, заторможенным. Отмечается снижение артериального давления и температуры тела, аритмия, тахикардия, выраженная бледность кожных покровов. Кожа больного краш-синдромом покрыта липким холодным потом. Возможна потеря сознания, непроизвольная дефекация и мочеиспускание. Иногда развивается отек легких. Уменьшается количество выделяемой мочи. Без адекватной врачебной помощи есть вероятность смертельного исхода в течение 1 или 2 суток.
На раздавленной конечности формируются очаги некроза. При отторжении мертвых тканей обнажаются мышцы, имеющие характерный вид вареного мяса. Развивается нагноение ран и эрозированных поверхностей. Появляется и постепенно нарастает острая почечная недостаточность. На 5-6 сутки у больных с синдромом длительного раздавливания развивается уремический синдром. Повышение уровня калия в крови вызывает аритмию и брадикардию.
На 5-7 сутки выявляются признаки легочной недостаточности. Нарастающая интоксикация, обусловленная поступлением в кровоток продуктов тканевого распада и бактериальных токсинов из раздавленной конечности, вызывает токсический гепатит. Возможен эндотоксический шок. Явления полиорганной недостаточности у пациентов с краш-синдромом постепенно уменьшаются в течение 2-3 недель.
Острая почечная недостаточность при краш-синдроме купируется примерно через месяц после травмы. Состояние пациента улучшается, температура его тела нормализуется. Уменьшаются боли и отек конечности. Некротизированные мышцы замещаются соединительной тканью, что приводит к атрофии мышц и развитию контрактур. При неблагоприятном развитии событий возможны местные (нагноение) и общие (сепсис) осложнения.
Диагностика
Патология диагностируется врачом-травматологом на основании характерного анамнеза (продолжительного сдавливания части тела), жалоб и данных внешнего осмотра. Для оценки общего состояния назначают комплекс лабораторных анализов. Для своевременного выявления и лечения ОПН осуществляют лабораторный мониторинг функции почек.
Лечение СДР
Перед освобождением конечности на нее необходимо наложить жгут выше места повреждения. После устранения сдавления конечность туго бинтуют и фиксируют на шине. Раны и поверхностные повреждения кожи обрабатывают по общим правилам. Пациенту с синдромом длительного раздавливания вводят наркотические анальгетики. Конечность обкладывают грелками со льдом. Если это возможно, выполняют футлярную новокаиновую блокаду раздавленной конечности или паранефральную блокаду по Вишневскому. Пострадавшего срочно доставляют в стационар.
Для улучшения микроциркуляции, борьбы с шоком и острой почечной недостаточностью в стационаре проводится инфузионная терапия (под контролем диуреза и центрального венозного давления). Для детоксикации и возмещения плазмопотери пациенту с краш-синдромом вводят 5% раствор глюкозы, солевые растворы, замороженную плазму, физ. раствор и раствор альбумина. Для улучшения микроциркуляции назначают гепарин (5000 ЕД) и реополиглюкин.
В целях компенсации метаболического ацидоза больному краш-синдромом капельно вводят 4% раствора гидрокарбоната натрия. Назначают антибиотики широкого спектра действия внутримышечно. Проводят симптоматическую терапию (диуретики, анальгетики, антигистаминные и противоаритмические препараты). При синдроме длительного раздавливания экстракорпоральную гемокоррекцию (гемодиализ, плазмаферез, плазмо- и гемосорбцию) проводят в как можно более ранние сроки.
При сохранении жизнеспособности мышечных тканей и выраженном субфасциальном отеке с нарушением местного кровообращения травматолог выполняет фасциотомию с ревизией и иссечением некротизированных мышечных пучков. Если нет нагноения, рану ушивают на 3-4 день, после уменьшения отека и улучшения общего состояния больного краш-синдромом.
В случаях необратимой ишемии проводят ампутацию конечности выше места наложения жгута. В других случаях показано иссечение некротизированных участков с сохранением жизнеспособных мышечных пучков. Жизнеспособность мышц определяют в ходе хирургического вмешательства. Критериями жизнеспособности является сохранение нормальной окраски, способность к кровоточивости и сокращению. После иссечения тканей рану обильно промывают антисептиками. Швы не накладывают. Рана заживает вторичным натяжением.
В отдаленном периоде больным с синдромом длительного раздавливания показаны курсы реабилитационного лечения (массаж, ЛФК), направленные на восстановление мышечной силы и устранение контрактур.
1. Диагностика и патогенетическое лечение синдрома длительного сдавления / Нечаев Э.А., Савицкий Г.Г. - 1992
3. Рекомендации ERBP по оказанию помощи пострадавшим с синдромом длительного сдавления при массовых катастрофах / перевод Камышовой Е.С. под ред.Захаровой Е.В. // Нефрология и диализ - 2015 - Т.17, №3
ДВС-синдром – расстройство гемостаза, связанное с гиперстимуляцией и дефицитом резервов свертывающей системы крови, приводящее к развитию тромботических, микроциркуляторных и геморрагических нарушений. При ДВС-синдроме наблюдается петехиально-гематомная сыпь, повышенная кровоточивость, дисфункция органов, а в острых случаях – развитие шока, гипотонии, сильных кровотечений, ОДН и ОПН. Диагноз устанавливают по характерным признакам и лабораторным тестам системы гемостаза. Лечение ДВС-синдрома направлено на коррекцию гемодинамики и нарушений свертывающей системы (антиагреганты, антикоагулянты, ангиопротекторы, гемотрансфузии, плазмаферез и др.).



Общие сведения
ДВС-синдром (диссеминированное внутрисосудистое свертывание, тромбогеморрагический синдром) – геморрагический диатез, характеризующийся чрезмерным ускорением внутрисосудистой коагуляции, образованием рыхлых сгустков крови в микроциркуляторной сети с развитием гипоксических и дистрофически-некротических измерений в органах. ДВС-синдром представляет опасность для жизни пациента из-за риска возникновения обширных, плохо купируемых кровотечений и острой дисфункции органов (главным образом, легких, почек, надпочечников, печени, селезенки), имеющих обширную микроциркуляторную сеть.
ДВС-синдром можно рассматривать, как неадекватную защитную реакцию, направленную на ликвидацию кровотечения при повреждении кровеносных сосудов и изоляцию организма от пораженных тканей. Встречаемость ДВС-синдрома в различных отраслях практической медицины (гематологии, реаниматологии, хирургии, акушерстве и гинекологии, травматологии и др.) достаточно велика.
Причины ДВС-синдрома
ДВС-синдром развивается на фоне заболеваний, протекающих с повреждением тканей, эндотелия сосудов и клеток крови, сопровождаемых микрогемодинамическими нарушениями и сдвигом гемостаза в сторону гиперкоагуляции. Основной причиной ДВС-синдрома выступают септические осложнения бактериальных и вирусных инфекций, шок любой природы. ДВС-синдром часто сопутствует акушерской патологии - тяжелому гестозу, предлежанию и преждевременной отслойке плаценты, внутриутробной гибели плода, эмболии амниотической жидкостью, ручному отделению последа, атоническим маточным кровотечениям, а также операции кесарево сечение.
Развитие тромбогеморрагического синдрома могут инициировать метастазирующие злокачественные опухоли (рак легкого, рак желудка), обширные травмы, ожоги, серьезные хирургические вмешательства. Нередко ДВС-синдром сопровождает трансфузию крови и ее компонентов, трансплантацию тканей и органов, протезирование сосудов и клапанов сердца, применение искусственного кровообращения.
Способствовать возникновению ДВС-синдрома могут сердечно-сосудистые заболевания, протекающие с гиперфибриногенемией, увеличением вязкости и снижением текучести крови, механическим препятствием кровотоку атеросклеротической бляшкой. К ДВС-синдрому могут приводить прием медикаментов (ОК, ристомицина, диуретиков), острые отравления (например, ядом змеи) и острые аллергические реакции.
Патогенез ДВС-синдрома
Несостоятельность гемостаза при ДВС-синдроме возникает за счет гиперстимуляции свертывающей и быстрого истощения антикоагулянтной и фибринолитической систем гемостаза.
Развитие ДВС-синдрома обуславливается различными факторами, которые появляются в кровяном русле и напрямую активируют процесс свертывания, либо осуществляют это через медиаторы, воздействующие на эндотелий. В качестве активаторов ДВС-синдрома могут выступать токсины, ферменты бактерий, околоплодные воды, иммунные комплексы, стрессовые катехоламины, фосфолипиды, снижение сердечного выброса и кровотока, ацидоз, гиповолемия и др.
Развитие ДВС-синдрома происходит с последовательной сменой 4-х стадий.
I - начальная стадия гиперкоагуляции и внутрисосудистой агрегации клеток. Обусловлена выбросом в кровь тканевого тромбопластина или веществ, обладающих тромбопластиноподобным действием и запускающих внутренний и внешний пути свертывания. Может продолжаться от нескольких минут и часов (при острой форме) до нескольких дней и месяцев (при хронической).
II - стадия прогрессирующей коагулопатии потребления. Характеризуется дефицитом фибриногена, кровяных пластинок и плазменных факторов вследствие их избыточного расхода на тромбообразование и недостаточного возмещения.
III - критическая стадия вторичного фибринолиза и выраженной гипокоагуляции. Отмечается разбалансировка гемостатического процесса (афибриногенемия, накопление патологических продуктов, разрушение эритроцитов) с замедлением свертывания крови (вплоть до полной неспособности к свертыванию).
IV - стадия восстановления. Наблюдаются либо остаточные очаговые дистрофические и некротические изменения в тканях тех или иных органов и выздоровление, либо осложнения в виде острой органной недостаточности.
Классификация ДВС-синдрома
По выраженности и скорости развития ДВС-синдром может быть острым (в т.ч., молниеносным), подострым, хроническим и рецидивирующим. Острая форма тромбогеморрагического синдрома возникает при массивном выбросе в кровь тромбопластина и ему подобных факторов (при акушерской патологии, обширных операциях, травмах, ожогах, синдроме длительного сдавления тканей). Характеризуется ускоренной сменой стадий ДВС-синдрома, отсутствием нормального защитного антикоагуляционного механизма. Подострая и хроническая формы ДВС-синдрома связаны с обширным изменением поверхности эндотелия сосудов (например, вследствие атеросклеротических отложений), выступающим в роли активирующей субстанции.
ДВС-синдром может проявляться локально (ограниченно, в одном органе) и генерализованно (с поражением нескольких органов или всего организма). По компенсаторному потенциалу организма можно выделить компенсированный, субкомпенсированный и декомпенсированный ДВС-синдром. Компенсированная форма протекает бессимптомно, микросгустки лизируются за счет усиления фибринолиза, факторы свертывания восполняются из резервов и путем биосинтеза. Субкомпенсированная форма проявляется в виде гемосиндрома средней степени тяжести; декомпенсированная - характеризуется каскадными реакциями реактивного фибринолиза, несостоятельностью коагуляционных процессов, несворачиваемостью крови.
ДВС-синдром может протекать с одинаковой активностью прокоагулянтного и сосудисто-тромбоцитарного звеньев гемостаза (смешанный патогенез) или с преобладанием активности одного из них.
Симптомы ДВС-синдрома
Клинические проявления ДВС-синдрома определяются темпом развития и распространенностью поражения, стадией процесса, состоянием компенсаторных механизмов, наслоением симптомов заболевания-индуктора. В основе ДВС-синдрома лежит комплекс тромбогеморрагических реакций и дисфункции органов.
При острой манифестной форме быстро (за несколько часов) развивается генерализованный ДВС-синдром, для которого характерно шоковое состояние с гипотонией, потерей сознания, признаками отека легких и острой дыхательной недостаточности. Гемосиндром выражается нарастающей кровоточивостью, массивными и профузными кровотечениями (легочными, маточными, носовыми, желудочно-кишечными). Характерно развитие очагов ишемической дистрофии миокарда, панкреонекроза, эрозивно-язвенного гастроэнтерита. Молниеносная форма ДВС-синдрома свойственна эмболии околоплодными водами, когда коагулопатия стремительно (в течение нескольких минут) переходит в критическую стадию, сопровождаясь кардиопульмонарным и геморрагическим шоком. Летальность матери и ребенка при этой форме ДВС-синдрома приближается к 80%.
Подострая форма ДВС-синдрома носит локальный характер с более благоприятным течением. Незначительный или умеренный гемосиндром проявляется петехиальной или сливной геморрагической сыпью, синяками и гематомами, усиленной кровоточивостью из мест инъекций и ран, кровотечениями из слизистых оболочек (иногда - «кровавый пот», «кровавые слезы»). Кожа приобретает бледный вид, мраморность, становится холодной на ощупь. В ткани почек, легких, печени, надпочечников, ЖКТ развиваются отек, резкое полнокровие, внутрисосудистая коагуляция, сочетание очагов некроза и множественных кровоизлияний. Самая распространенная - хроническая форма ДВС-синдрома часто имеет бессимптомное течение. Но по мере прогрессирования фонового заболевания нарастают проявления геморрагического диатеза и нарушения функции органов.
ДВС-синдром сопровождается астеническим синдромом, плохим заживлением ран, присоединением гнойной инфекции, развитием келоидных рубцов. К осложнениям ДВС-синдрома относятся гемокоагуляционный шок, острая дыхательная недостаточность, ОПН, некроз печени, язвенная болезнь желудка, инфаркт кишечника, панкреонекроз, ишемический инсульт, острая постгеморрагическая анемия.
Диагностика ДВС-синдрома
Для установления ДВС-синдрома необходимы тщательный сбор анамнеза с поиском этиологического фактора, анализ клинической картины и данных лабораторных исследований (общего анализа крови и мочи, мазка крови, коагулограммы, паракоагуляционных проб, ИФА). Важно оценить характер кровоточивости, уточнить стадию коагулопатии, отражающую глубину нарушений.
Для ДВС-синдрома характерна петехиально-гематомная кровоточивость, геморрагии сразу из нескольких мест. При малосимптомном течении гиперкоагуляция выявляется только лабораторными методами. К обязательным скрининговым тестам относятся определение количества тромбоцитов, фибриногена, АПТВ, протромбинового и тромбинового времени, времени свертывания по Ли-Уайту. Исследование маркеров внутрисосудистого свертывания - РФМК и ПДФ, D-димера методом ИФА и паракоагуляционных проб помогает подтвердить ДВС-синдром.
Критериями ДВС-синдрома являются наличие фрагментированных эритроцитов в мазке крови, дефицит тромбоцитов и фибриногена, повышение концентрации ПДФ, падение активности антитромбина III в сыворотке крови, удлинение АПТВ и тромбинового времени, отсутствие образования или нестабильность сгустка или in vitro. Выполняется оценка функционального состояния «шоковых органов»: легких, почек, печени, сердечно-сосудистой системы, головного мозга. ДВС-синдром необходимо дифференцировать от первичного фибринолиза, других коагулопатических синдромов.
Лечение ДВС-синдрома
Успех лечения ДВС-синдрома возможен при его ранней диагностике. Активные лечебные мероприятия требуются при выраженной симптоматике в виде кровотечений и органной недостаточности. Больных с ДВС-синдромом следует госпитализировать в ОРИТ и при необходимости проводить ИВЛ, активную противошоковую терапию. При малосимптомном ДВС-синдроме основным представляется лечение фоновой патологии, коррекция параметров гемодинамики и функциональных нарушений органов.
Острый ДВС-синдром требует срочного устранения его первопричины, например, экстренного родоразрешения, гистерэктомии - при акушерской патологии или антибиотикотерапии – при септических осложнениях. Для ликвидации гиперкоагуляции показано введение антикоагулянтов (гепарина), дезагрегантов (дипиридамола, пентоксифиллина), фибринолитиков. Больные должны находиться под постоянным динамическим контролем показателей гемостаза.
В качестве заместительной терапии при ДВС-синдроме применяются трансинфузии свежезамороженной плазмы, тромбоцитарной или эритроцитарной массы (при падении уровня тромбоцитов или Hb); криопреципитата (при сердечной недостаточности), физраствора. При кровотечениях, угрожающих жизни, возможно назначение антифибринолитических средств (аминокапроновой к-ты, ингибиторов протеаз). При кожных геморрагиях и ранах накладываются повязки с этамзилатом, гемостатическая губка.
По показаниям применяют кортикостероиды, оксигенотерапию, плазмаферез. Для восстановления микроциркуляции и нарушенных функций органов назначают ангиопротекторы, ноотропные препараты, посиндромную терапию. В случае ОПН проводят гемодиализ, гемодиафильтрацию. При хроническом ДВС-синдроме целесообразно использование дезагрегантов, вазодилататоров, в послеоперационном периоде - гепаринотерапии.
Прогноз и профилактика ДВС-синдрома
Прогноз ДВС-синдрома - вариативный, зависит от основного, этиологически значимого заболевания, тяжести нарушений гемостаза и своевременности начатого лечения. При остром ДВС-синдроме не исключен летальный исход в результате некупируемой большой кровопотери, развития шока, ОПН, острой дыхательной недостаточности, внутренних кровоизлияний. Предупреждение ДВС-синдрома заключается в выявлении пациентов группы риска (особенно, среди беременных и лиц пожилого возраста), лечении фонового заболевания.
Синдром Драве – это детская энцефалопатия наследственного характера, которая характеризуется эпилептиформными приступами, отставанием в психическом развитии и резистентностью к противоэпилептической терапии. Клинически заболевание проявляется полиморфными эпилептическими припадками, неврологическими расстройствами, атипическими абсансами и фокальными моторными пароксизмами. Диагностика синдрома Драве базируется на характеристике возникающих приступов, данных ЭЭГ и МРТ, идентификации мутации генов SCN1A или GABRG2. Лечение малоэффективно и проводится с целью уменьшения частоты приступов, профилактики эпилептического статуса.
Общие сведения
Синдром Драве или тяжелая миоклоническая эпилепсия младенчества – это аутосомно-доминантная энцефалопатия с дебютом в первые 12 месяцев жизни ребенка, которая проявляется фебрильными и афебрильными генерализованными приступами, фокальными миоклоническими пароксизмами, расстройствами неврологического статуса и дефицитом интеллекта. Впервые заболевание было описано французским психиатром и эпилептологом Шарлоттой Драве в 1978 году. Встречается данный синдром редко, распространенность – 1:20-40 тысяч детского населения. У мальчиков патология возникает вдвое чаще, чем у девочек. Исход синдрома Драве неблагоприятный – заболевание неизлечимо и слабо поддается медикаментозной терапии. Летальность составляет порядка 16-18%.
Причины и симптомы синдрома Драве
Синдром Драве – это генетически детерминированная патология, которая передается по аутосомно-доминантному типу наследования. Спровоцировать развитие тяжелой миоклонической эпилепсии младенчества могут мутации локуса SCN1A на 24 участке длинного плеча 2 хромосомы (в 80% случаев) или GABRG2 на 5q34. Данные гены кодируют α1-субъединицу Na+-каналов, что приводит к нарушению физиологических процессов реполяризации и деполяризации в нейронах, и как следствие – к патологической активности ЦНС.
В клинической картине синдрома Драве выделяют 3 этапа развития: фебрильный (до 12-24 месяцев), агрессивный или катастрофический (2-8 лет), статический (старше 8 лет). Дебют заболевания происходит в возрасте от 2 месяцев до 1 года, в среднем – в 5 месяцев. До момента возникновения первых симптомов ребенок развивается нормально, неврологических и психических отклонений не наблюдается. В большинстве случаев первичными проявлениями фебрильной стадии синдрома Драве становятся фибриллярные судороги атипического характера. Они имеют большую продолжительность (свыше 20 минут), включают в себя очаговые компоненты и альтернирующие гемиконвульсии, иногда переходят в эпилептический припадок. На ранних этапах такие состояния сопровождаются субфебрильной или фебрильной температурой тела, в дальнейшем подобных проявлений не наблюдается. Зачастую при синдроме Драве приступ может быть спровоцирован гипертермией (согреванием, горячей ванной или инфекционной патологией), световыми раздражителями, резкими движениями и т. д.
Катастрофический или агрессивный период синдрома Драве характеризуется выраженными полиморфными клонико-тонико-клоническими припадками, альтернирующими гемиконвульсиями, очаговыми моторными пароксизмами, атипичными абсансами. Приступы начинаются с мышечных подергиваний по всему телу (иногда – асинхронных), переходят в кратковременную тоническую, а затем – клоническую фазы. Часто подобное состояние трансформируется в эпилептический статус, который может сохраняться до нескольких суток. В возрасте 1-2 лет у больных с синдромом Драве определяется дефицит интеллекта (олигофрения) и гиперактивность, поведенческие аномалии, нарастающие до 6-7 лет и сохраняющиеся на протяжении всей жизни. Также развиваются неврологические нарушения: мышечная гипотония, атаксия, интенционный тремор, моторная неловкость, признаки пирамидной недостаточности. В этом же возрасте у части детей возникает паттерн-сенситивность, при которой определенная одежда, обои или телевизионные передачи могут стать причиной очередного приступа.
Статическая стадия синдрома Драве характеризуется уменьшением интенсивности и частоты эпилептических припадков. Психические и неврологические отклонения остаются. Большая часть приступов возникает в ночное время или сразу после пробуждения. Как и в других периодах, они могут быть спровоцированы повышением температуры тела, ярким светом, резким движением и др. На фоне отставания в интеллектуальном развитии, нарушений психики и резистентности заболевания к лечению пациент почти полностью лишен способности адаптироваться в социуме.
Диагностика синдрома Драве
Диагностика синдрома Драве основывается на анамнестических данных, физикальном обследовании, лабораторных и инструментальных методах исследования. Из анамнеза педиатром выясняется возраст, в котором произошла манифестация патологии, первичные проявления, характеристика приступов, степень их тяжести и динамика развития. При осмотре ребенка в межприступный период можно выявить отставание в интеллектуальном развитии (ЗПР), гиперактивность, нарушения неврологического статуса. Во время припадка определяются атипичные абсансы, очаговые расстройства, альтернирующие гемиконвульсии.
Общие лабораторные анализы (ОАК, ОАМ, анализ кала) малоинформативны – выраженные отклонения от возрастной нормы, как правило, отсутствуют. Из инструментальных методов исследования при синдроме Драве используются электроэнцефалограмма (ЭЭГ) и магнитно-резонансная томография (МРТ). Между приступами на ЭЭГ у большинства таких детей определяется сочетание очаговой, мультирегиональной и диффузной эпилептиформной активности с нарастанием во сне. При низкой частоте припадков данные признаки могут отсутствовать. По результатам МРТ головного мозга удается установить признаки диффузной атрофии коры головного мозга и мозжечка, субкортикальных слоев, иногда – увеличение размеров желудочков. Для подтверждения синдрома Драве используется кариотипирование с определением мутации генов SCN1A или GABRG2.
В педиатрии дифференциальная диагностика синдрома Драве проводится с фебрильными судорогами, митохондриальными и дисметаболическими патологиями, доброкачественной миоклонической эпилепсией младенчества, синдромами Леннокса-Гасто и Дозе, другими формами эпилепсии у детей, которые сопровождаются миоклоническими припадками. Практически идентичную клиническую картину имеет мутация гена PCDH19 – эпилепсия с умственной отсталостью, ограниченная женским полом.
Лечение синдрома Драве
Синдром Драве – это форма эпилепсии у детей, которая почти не поддается терапии. Основная цель лечения – снизить чистоту приступов, профилактировать их трансформацию в эпилептический статус. Как правило, большинство распространенных противоэпилептических средств при тяжелой миоклонической эпилепсии младенчества неэффективны. В качестве стартовой терапии показаны вальпроаты (вальпроева кислота) и сульфат-замещенные моносахариды (топирамат). Также могут применяться фармакологические средства из групп барбитуратов и бензодиазепинов. В некоторых случаях при синдроме Драве позитивная динамика отмечается на фоне кетогенной диеты, которая подразумевает большое количество жиров и строгое ограничение углеводов.
Прогноз и профилактика синдрома Драве
Прогноз для жизни при синдроме Драве сомнительный, для выздоровления – неблагоприятный. Дефицит интеллекта, расстройства психики, эпилептические припадки и неврологические нарушения обычно сохраняются на протяжении всей жизни человека, что обусловливает его полную социальную дезадаптацию. Обычно приступы возникают в ночное время или сразу после пробуждения, а их интенсивность и частота уменьшаются. Смертность составляет порядка 15,9-18%. Основные причины – синдром внезапной детской смерти при эпилепсии, интеркуррентные инфекционные заболевания, несчастные случаи во время припадков.
Антенатальная профилактика синдрома Драве аналогична другим наследственным заболеваниям. Она подразумевает медико-генетическое консультирование и планирование беременности, кариотипирование плода посредством амнио- или кордоцентеза. Постнатальные превентивные меры включают в себя исключение гипертермических состояний у ребенка (раннее лечение инфекционных заболеваний, избегание горячих ванн и т. д.) и других факторов, которые могут спровоцировать приступ.

Синдром Драве – это редкая, катастрофическая, пожизненная форма эпилепсии, которая начинается на первом году жизни с частых и / или длительных припадков. Когнитивные, поведенческие проблемы начинаются в возрасте 2-3 лет.
Синдром Драве был впервые описан доктором Шарлоттой ДРАВЕТ как « тяжелая миоклоническая эпилепсия младенчества», но с тех пор был признан во всем мире.
Первое описание клинической картины было:
Приступы появились в возрасте до одного года у нормально развивающегося ребенка. Первыми приступами были судороги (клонические или тонико-клонические), связанные с лихорадкой. Эти припадки часто были длительными или очень длительными (в некоторых случаях более одного часа) и требовали неотложной терапии (ректальное или внутривенное введение противосудорожного препарата).
В возрасте от 2 до 3 лет развивались другие типы судорог (миоклонические судороги, атипичные отсутствия, фокальные (очаговые) судороги,
сопровождающиеся регрессом развития или с нарушениями поведения. КТ головного мозга оставалась нормальной. Поскольку все эти клинические признаки были не у каждого пациента, критерии были расширены, и в 2001 году Международная по борьбе с эпилепсией изменила название тяжелой миоклонической эпилепсии младенчества на «синдром Драве». Точная частота в общей популяции неизвестна. В 1990 году было подсчитано, что синдром Драве возникал у одного из 1/20 000 и 1/40 000 рождений. Вероятно, эта частота была недооценена, потому что болезнь была недостаточно известна в то время. Мальчики примерно в два раза чаще девочек болеют синдромом Драве.
Симптомы синдрома Драве
Судороги являются самым ранним симптомом синдрома Драве, они начинаются на первом году жизни , чаще в первые 5 месяцев жизни ребенка, до этого ребенок неврологически полностью здоров и не отстает в развитии от сверстников. Задержка психоречевого развития, а также судороги, как правило,усугубляются по мере взросления ребенка.
Читайте также: